
The membrane environment is a key determinant of function and/or organization of membrane proteins. It is essential for the activities of the Consortium to have access to quantitative information about the characteristics and energetics of membrane-protein interactions. For multi-segment transmembrane proteins in particular, such characterization is complicated by the different hydrophobic thicknesses of the component transmembrane segments and the radially non-uniform hydrophobic interface created between the membrane and the protein. A hybrid Continuum-Molecular Dynamics (CTMD) approach to compute membrane deformations profiles around multi-segment proteins and corresponding energetics was recently published [Sayan Mondal, George Khelashvili, Jufang Shan, Olaf Andersen, Harel Weinstein (2011), Biophysical J (101): 2092-2101; link]. This stand-alone application implements the CTMD approach.
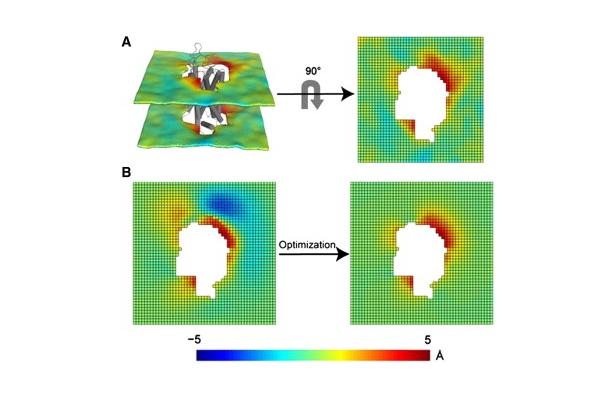 Protocol for the 3D-CTMD approach, illustrated for rhodopsin in a diC14:1PC lipid bilayer (Mondal et al., BJ 2011). Click to enlarge and learn more.
Protocol for the 3D-CTMD approach, illustrated for rhodopsin in a diC14:1PC lipid bilayer (Mondal et al., BJ 2011). Click to enlarge and learn more.The CTMDapp software calculates the deformation profiles of the bilayer and the free energy cost of the membrane deformation around multi-segment transmembrane proteins, taking into account the radially non-uniform hydrophobic surface of the protein. As the primary input it requires a molecular dynamics trajectory of a multi-segment, transmembrane protein embedded in a bilayer.
To allow for calculations without a molecular dynamics input, the CTMDapp software also implements a simplified version of the CTMD method that can assess the radial asymmetry of the membrane-protein interface for a particular protein structure at an approximate level.
Methodological details can be found in the original paper (Mondal et al., BJ 2011). In addition, the application is documented with brief usage notes at every step and generates diagnostic intermediate output.
Download the software:
- Windows: http://memprotein.org/software/CTMDapp_windows_pkg.exe
- Mac: http://memprotein.org/software/CTMDapp_mac_pkg.zip
- Unix: http://memprotein.org/software/CTMDapp_unix_pkg.zip
Instructions:
For the Windows version, clicking on the downloadable .exe file will directly launch the Matlab Component Runtime (MCR) installation process and extract the app executable. For the Mac version, the MCR must be installed first by opening MCRinstaller.dmg in the folder. This is a one-time installation, after which CTMDapp can be launched. The Unix version requires an interface like Xwin along with a script setting proper paths.
The software was written by Sayan Mondal ([email protected]) in the Harel Weinstein ([email protected]) lab at Weill Cornell Medical College, Cornell University. Please feel free to contact us with questions and suggestions about the software.






Dear:
I am using gromacs for membrane protein simulation. If possible, would you please tell me how to use CTMD for Gromacs output analysis? I am preparing a manuscript for my work and I will cite CTMD.
thank you very much.
Shuguang
Hi Shuguang,
I suggest that you simply convert your Gromacs output (xtc?) to dcd format. I have replied to your email, and we can go back and forth by email till it works for you.
Sayan
Hi,
Would CTMD also work with coarse-grained trajectories (run in the gromacs environment)?
Thx
Markus
Yes, I have used it with CG trajectories run in Gomacs as well. The trajectory has to be converted to dcd format, and then use the phosphate beads. If you face any problem, please do email me.
Sayan
Hello,
I have carry out several 3D-CTMD calculations. Unfortunately I am not able to obtain the files which contain the coordinates of the optimized grids. Is it possible to obtain that output?
Thanks in advance
Yes, the optimized results are in the output Matlab file, whose filename ends with “_results.mat”. You will find the results stored in the variable u2_optimal, if you load this .mat file in Matlab. Alternatively, you can save the figure as a .fig Matlab figure, and extract the data in Matlab. In the output figures, you can also query the value at specific gridpoints using the data cursor (+ symbol). Finally, you can make Matlab figures from the results.mat file using the CTMDapp software, by starting the software and then going to “View results from completed energy calculations”.
Does this answer your needs?
Hi,
Can you give all the details on how to install the CTMD application on the Linux system.
Regards,
Namita
Hi,
After downloading the Unix package, please make sure that the files have the correct permissions and install the Matlab Compiler Runtime (MCR) by clicking on MCRInstaller.bin. You may also have to do a dos2unix conversion for the executable .sh file. Then you can run the executable .sh file from the command line with the argument specifying the path for the MCR — details are in readme.txt. By the way, note that the MCR gets installed in a folder named v711 (has to do with the version number). I hope this helps, and please let me know if you need any further help.
Hi,
Thanks for your reply.
I have simply convert my Gromacs (membrane-protein) output (xtc) to dcd format using vmd .Now whenever I am trying to run this ./run_CTMDapp_unix.sh /home/nd346/Software/CTMD/v711 it runs the application but when I specify the dcd file it runs for a while and then terminate with the following error:
”
??? Attempted to access avgzu(:,1); index out of bounds because
size(avgzu)=[0,0].
Error in ==> thickness_calculate at 3
Error in ==> ctmd_ver2 at 204
MATLAB:badsubscript
Warning: 2 visible figure(s) exist at MCR termination.
If your application has terminated unexpectedly, please note that
applications generated by the MATLAB Compiler terminate when there are no
visible figure windows. See the documentation for WaitForFiguresToDie and
WAITFORCALLBACKS for more information.
Warning: Class
‘uitools.uimodemanager’
in use at MCR termination.
If your application has terminated unexpectedly, please note that
applications generated by the MATLAB Compiler terminate when there are no
visible figure windows. See the documentation for WaitForFiguresToDie and
WAITFORCALLBACKS for more information.
Warning: Class
‘uitools.uimode’
in use at MCR termination.
If your application has terminated unexpectedly, please note that
applications generated by the MATLAB Compiler terminate when there are no
visible figure windows. See the documentation for WaitForFiguresToDie and
WAITFORCALLBACKS for more information.
Warning: Class
‘graphics.datacursormanager’
in use at MCR termination.
If your application has terminated unexpectedly, please note that
applications generated by the MATLAB Compiler terminate when there are no
visible figure windows. See the documentation for WaitForFiguresToDie and
WAITFORCALLBACKS for more information.
”
Can you suggest what is wrong with it.
waiting for your reply,
Regards,
Namita
Hi Namita,
Does your input dcd file contain the protein? The input dcd file should contain only the Phosphate beads.
Hi,
I am trying to use CTMD for getting a continuum model of the trajectory I got from a NAMD simulation. My model contains a protein (ion channel) embedded in a lipid bilayer. As emphasized in the software manual, I have used the dcd file which only contain the Phosphate molecules of the lipid as an input file. Then the program prompts first for the number of lipids and a name for the output. However, as soon as I enter these info, the program crashes without any error or output.
FYI, I am using windows 7 and I have Matlab R2003b pre-installed on my PC (in addition to the Matlab compiler) accompanied by CTMD.
Am I missing something?
Many thanks in advance,
Navid
P.S. I do have to open CTMD several time to be able to input my file before crashing but even when I can input the dcd file I still have the above-mentioned issue.
Hi Navid, could you please email me the log file? Windows 7 is fine, but is there a chance that this could be a memory issue with your computer?
Sayan
Hi Sayan,
I have a doubt regarding the significance of the values of delta G provided by CTMDapp. The final results are two values of G. One of them is the result of the minimization of the deformed membrane and the other concerns the penalty energy of a flat membrane, aren’t they?
Then, I am not sure if the delta G value i have to use is just the value corresponding to the deformed system or, maybe the difference between this one and the other corresponding to the flat membrane.
I would say that the result of the minimization is the difference between the penalty of the deformed membrane and the undeformed one which accounts for spontaneous curvature and this value will be always > 0.
In turn, i think the flat membrane value is the energy of an unmeaningfull membrane which does not account for these spontaneous curvature and it is sometimes greater then the penalty of the deformed system. Then the difference between deformed and flat is <0 but does not make sense as the value of the deformation penalty. Then the membrane would deform in this way in the absence of inclusion, wouldn't it?
After trying to explain the mess in my brain, could you help me to understand which value should I consider as the correct deformation energy penalty?
Thank you in advance.
john